安徽师范大学代胜瑜教授课题组J. Catal.文章:金属-π相互作用下杂原子二苯并环庚基对乙烯聚合链转移与链行走的调控研究
发表时间 :2024-05-22
作者 :高分子科学前沿
来源 :高分子科学前沿
链行走与链转移是决定通过后过渡金属催化乙烯(共)聚合所生产聚烯烃结构和分子量的两大核心因素。这些过程受多重因素影响,既包括外部聚合条件,例如助催化剂类型、温度、单体浓度及链转移试剂,也涵盖催化活化位点周边的配体环境,特别是配体的空间位阻和电子效应。在诸多因素中,配体空间位阻在后过渡金属催化体系中起到了关键的调节作用,用以平衡链行走、链转移及链增长之间的关系。研究显示,在多数后过渡金属催化体系中,增大催化中心的轴向空间位阻能有效抑制链转移,进而在相同条件下生产出分子量更高的聚合物。例如,一系列不对称二芳基甲基α-二亚胺镍和钯催化剂(图1a-b)的空间位阻逐步增加时,所得聚乙烯和共聚物的分子量也随之逐步增加。然而,这种趋势并非放之四海而皆准,因为链增长同样可能受到空间位阻的制约。在某些情境下,链增长受到的抑制可能更为显著,导致所得聚合物的分子量反而低于那些由弱屏蔽催化中心生产的聚合物。例如,某些不对称α-二亚胺镍催化剂能迅速产出高分子量聚乙烯。同样地,空间位阻的变化也可能影响链行走过程及所得聚合物的微观结构,诸如支化度的改变。先前的研究揭示,随着钯催化中心空间位阻的增加,所得聚乙烯和共聚物的支化度逐渐降低(Macromolecules 2016, 49, 8855–8862)。相反地,在相应的镍催化体系中,聚乙烯的支化度最初随镍催化中心空间位阻的增加而上升,随后又出现下降(Organometallics 2019, 38, 2919-2926)。这些发现凸显了通过调整配体空间位阻,我们能够定制出具有理想分子量和支化度的聚合物。经过精心调控,我们可以改变聚烯烃的结构与性能,从而制备出目标聚合物。
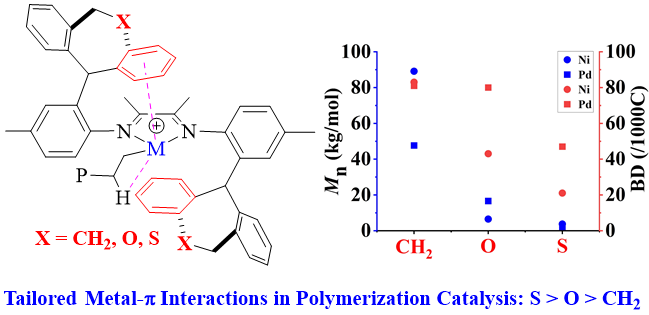
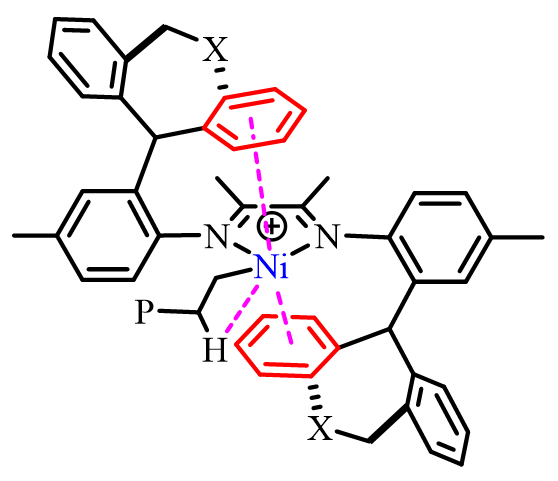
此外,调节配体的电子效应也有可能在一定程度上影响所得聚合物的分子量、支化度和拓扑结构,这一点在中性水杨醛亚胺镍体系中尤为明显。例如,Mecking等人报道的N-三苯基水杨醛亚胺镍催化剂中,具备吸电取代基(如CF3、SF5、NO2)的催化剂能聚合得到高分子量线性聚合物,而具备给电子取代基(如Me、OMe)的催化剂则在相同条件下聚合得到超支化低分子量聚合物(图1c)(Acc. Chem. Res. 2020, 53, 2738–2752)。理论计算与实验证据均表明,邻芳基芳环与镍中心之间较弱的相邻基团相互作用有利于β-H的消除,进而促进链转移与支化形成。近期的一些报道还发现了类似的弱邻基相互作用,涉及[N^N]体系中阳离子镍和钯的远程非共轭电子效应(图1d)( Polym. Chem. 2020, 11, 2692–2699)及第二层配位空间效应(图1e)( Angew. Chem., Int. Ed. 2017, 56, 11604–11609)。尽管已观察到一些有意义的模式,但这些相互作用对聚合活性、分子量及所得聚合物微观结构的影响尚不十分明确。在本研究中,我们通过用具有不同给电子能力的杂原子取代催化剂中桥接的亚乙基碳原子,从而实现了对所得(共)聚合物分子量的精确调控(图1f)。值得注意的是,我们观察到弱金属-π相互作用实际上是抑制而非促进链行走,这与先前报道的趋势形成了鲜明对比。
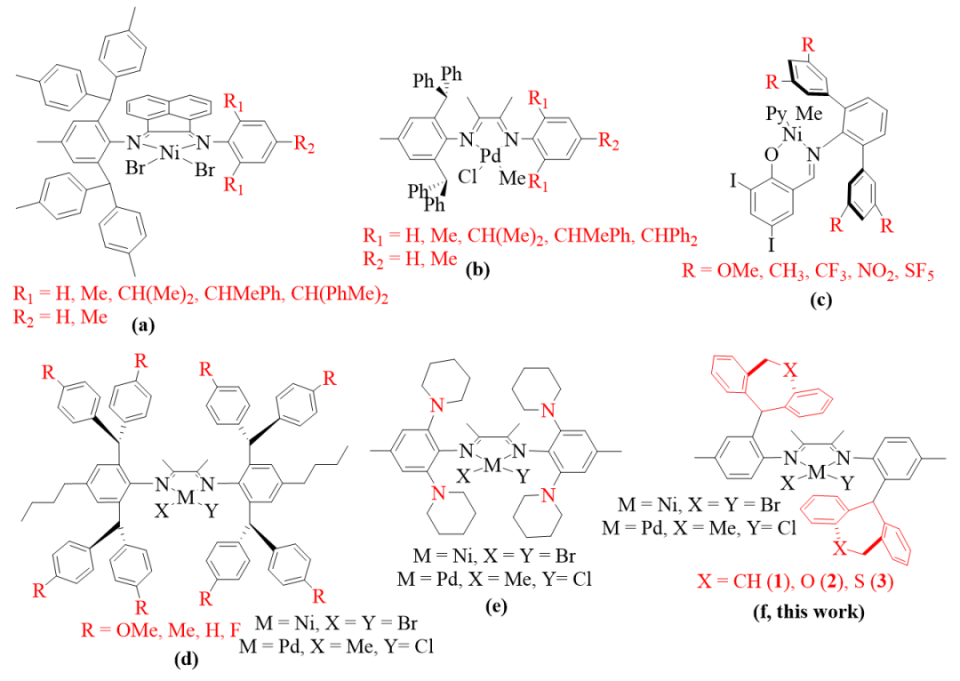
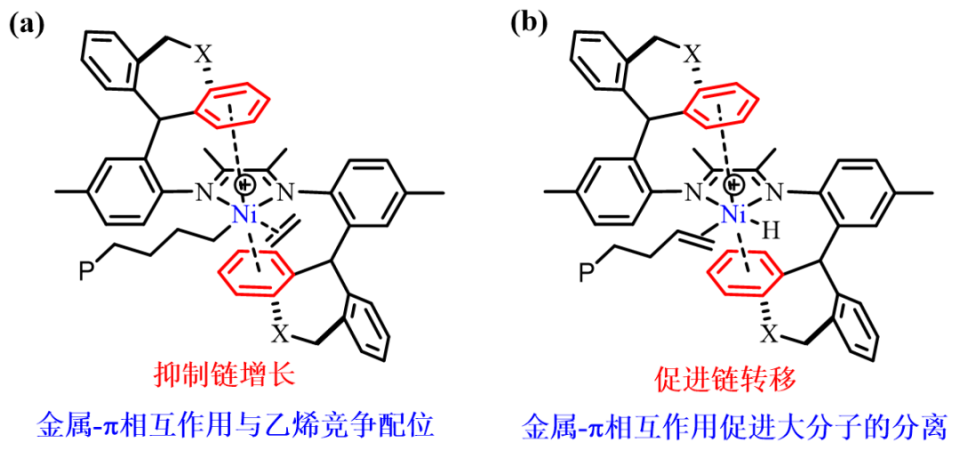
图1. 配体空间位阻(a-b)和电子效应(c-f)修饰的后过渡金属催化剂
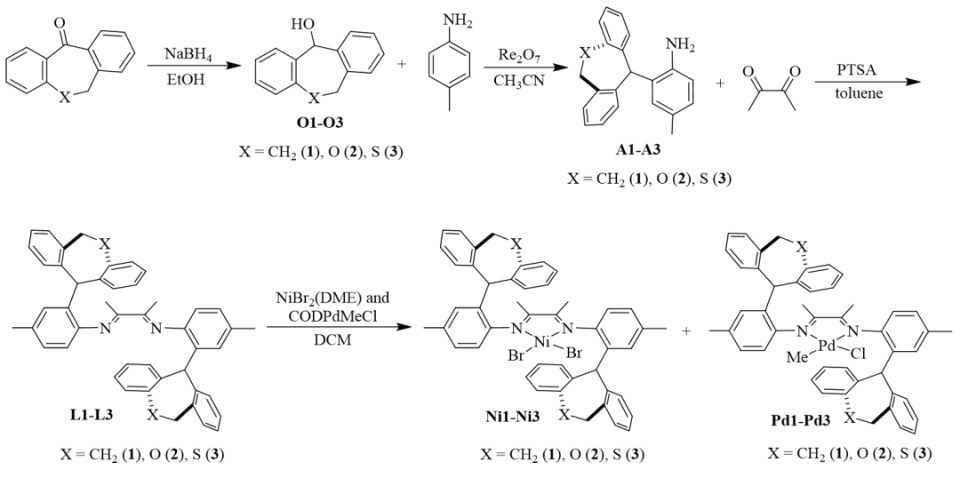
图2. 含二苯并环庚基及杂原子取代的二苯并环庚基α-二亚胺配体和其相应的镍和钯催化剂
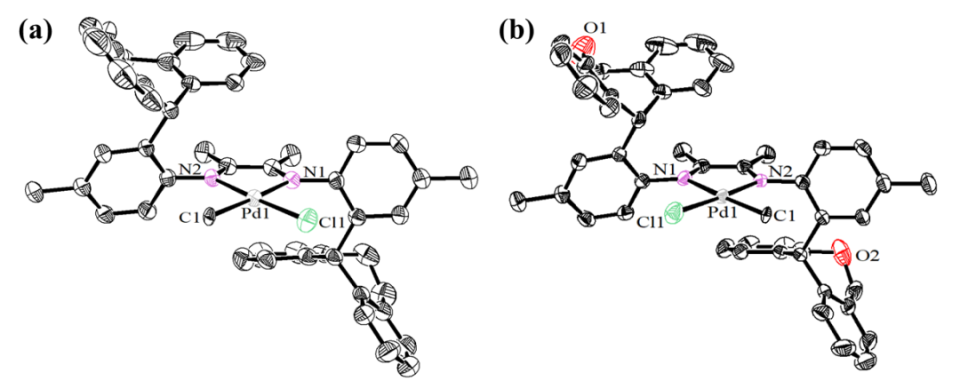
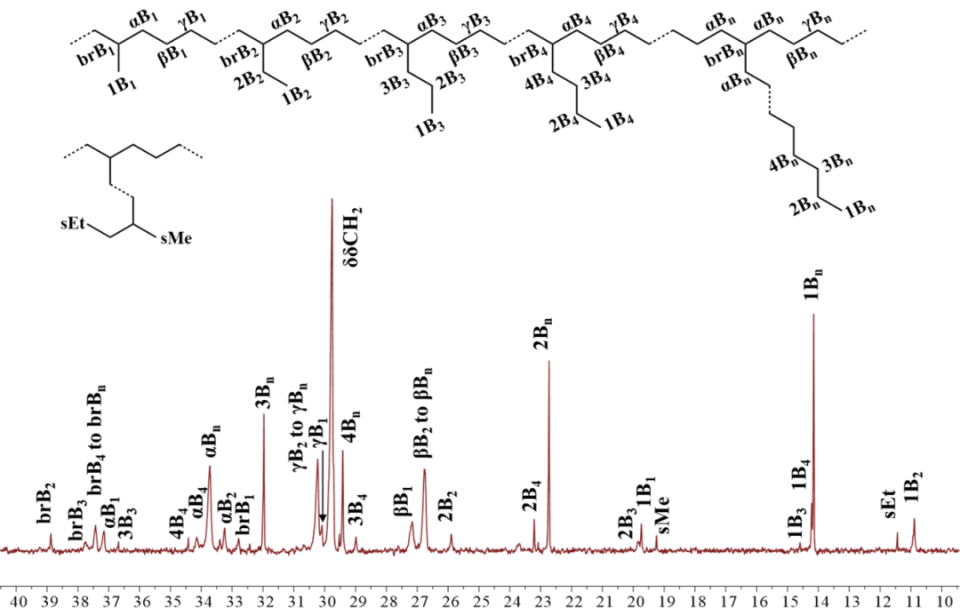
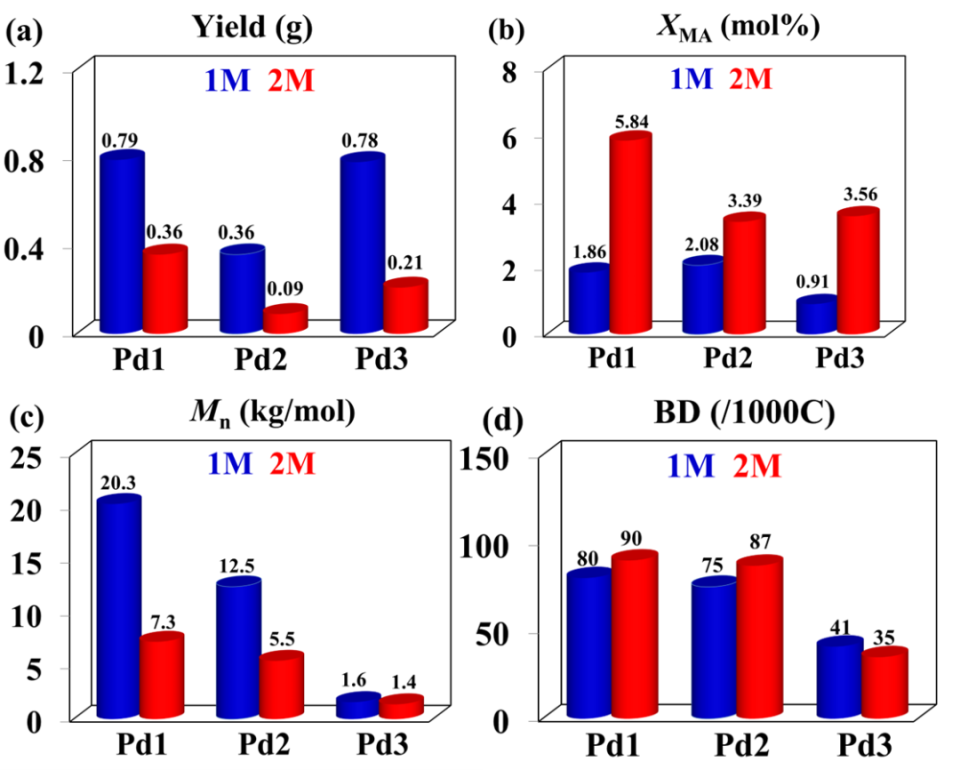
按照先前报道的方法,我们采用NaBH4还原对应的二苯并庚酮,成功合成了二苯并庚醇及其杂原子取代的醇类(O1-O3)。紧接着,我们通过金属催化的Friedel-Crafts烷基化反应,进一步制备了单侧苯并环庚基苯胺及其含杂原子的取代胺类(A1-A3)。之后,在对甲苯磺酸的催化作用下,我们将苯胺A1-A3与2,3-丁二酮在120 ℃的甲苯环境中进行缩合,从而得到了α-二亚胺配体L1-L3(详见图2)。为了确保化合物的准确性,我们使用了1H和13C NMR以及高分辨率质谱(HRMS)对所有新合成的化合物进行了详尽的表征。当α-二亚胺配体(L1-L3)与1当量的(DME)NiBr2或(COD)PdMeCl反应时,能够生成相应的催化剂Ni1-Ni3和Pd1-Pd3,且产率分别达到了59-74%和52-70%(参见图2)。我们对镍催化剂进行了元素分析表征,同时,对钯催化剂则采用了更为全面的1H和13C NMR、元素分析以及X射线单晶衍射进行表征。为了进一步探究催化剂的结构,我们通过乙醚对催化剂的二氯甲烷溶液进行分层,成功获得了催化剂Pd1和Pd2的单晶,并对其进行了X射线衍射分析。分析结果显示,这两种钯催化剂在Pd金属中心周围均呈现出平面正方形几何结构(见图3)。从单晶结构中我们可以清晰地观察到,二苯并环庚基取代基对钯中心的轴向位置起到了有效的屏蔽作用。特别是在催化剂Pd2的单晶中,两个氧原子与金属中心之间的距离(Pd1-O1=6.31Å,Pd1-O2=6.87Å)远大于两个原子间的范德华半径总和(3.15Å),这表明氧原子与金属中心之间直接相互作用的可能性极低。相较之下,二苯并环庚基取代基中的苯基中心与金属中心的距离仅为3.95 Å,这揭示了在聚合过程中可能存在弱相互作用,因为这个距离已接近金属-π相互作用的临界值。此外,系统中的杂原子能够通过调节苯环上的电子密度来影响与金属中心的相互作用强度,这种调节机制为控制乙烯聚合过程中的链转移和链行走提供了有效手段。经过200当量助催化剂Et2AlCl的活化处理后,三种镍催化剂在乙烯聚合反应中均展现出了卓越的活性。在相同的实验条件下,这三种催化剂的活性排序为Ni1>Ni2>Ni3,其中Ni1的活性显著高于其他两种催化剂(见图4a)。Ni2和Ni3活性的降低,可能是由于杂原子取代的二苯并环庚基与催化中心之间产生了更强的金属-π相互作用,这种作用抑制了链的增长过程(见图5a)。特别值得注意的是,含有硫的取代基对催化剂活性的抑制作用更为显著。随着温度从30℃升高到50℃,Ni1和Ni3的聚合活性逐渐增强,但在70℃时活性开始下降。与此不同,Ni2的聚合活性则随着温度的升高而持续下降(见图4a)。这种变化可以归因于催化剂稳定性和活性随温度变化的相互作用。金属-π相互作用不仅对催化剂的活性有影响,还显著改变了所得聚乙烯的分子量和微观结构。由Ni1催化制得的聚乙烯,其分子量明显高于由其他两种催化剂制得的聚乙烯,差距超过一个数量级(见图4b)。在乙烯聚合过程中,富电子的杂原子取代二苯并环庚基通过与双键中间体竞争配位到金属中心,从而促进了链转移过程。这种促进作用随着取代基电子丰度的增加而增强(见图4b和图5b)。聚乙烯的分子量主要由链增长速率与链转移速率的比值决定。由于金属-π相互作用在加速链转移的同时减缓了链增长,因此这一比值显著降低,导致聚乙烯的分子量大幅下降。同时,随着温度的升高,所得聚乙烯的分子量逐渐降低(见图4b),这一观察结果与先前关于α-二亚胺镍体系的报道相一致,即温度升高会导致链转移与链增长的比率上升。最引人注目的是,这种金属-π相互作用对聚乙烯的支化度也产生了显著影响。如图4c所示,与Ni2(43-48/1000C)和Ni3(21-32/1000C)催化合成的聚乙烯相比,Ni1(83-90/1000C)催化合成的聚乙烯具有明显更高的支化度。富电子的杂原子取代二苯并环庚基通过抑制乙烯聚合过程中的β-H消除来阻碍链行走,从而降低聚乙烯的支化度(见图6)。特别是更富电子的硫代二苯并环庚基取代基,对链行走的抑制作用更为显著,导致聚乙烯的支化度最低(见图4c和图6)。此外,根据先前对α-二亚胺镍体系的研究,温度升高会增加聚乙烯的支化度,因为高温条件更有利于链行走而非链增长。这些支化聚乙烯的熔点各不相同,许多样品甚至表现出双熔点特性。这种现象可能是由多种因素共同作用的结果,如聚合物分子量的降低以及非均质支化结构等,这些因素都可能导致聚合物的结晶过程变得不规则。图4. 在30-70 ℃条件下,Ni1-Ni3生成聚乙烯的产量(a)、分子量(b)和支化度(c)图5. 富电子杂原子取代二苯并环庚基取代基促进链转移发生的可能机制图6. 富电子的杂原子二苯并环庚基与β-H竞争与金属中心的相互作用抑制β-H消除的反应机制经过2.0当量的四(3,5-二(三氟甲基)苯基)硼酸钠(NaBArF)的原位活化后,钯催化剂展现出了中等活性。在30℃时,装载了含杂原子二苯并环庚基取代基的催化剂Pd2和Pd3的活性超过了Pd1。然而,当温度升高到50℃时,这一活性趋势发生了反转(图7a)。与镍体系相比,钯体系中的反应情况更为复杂,这种复杂性可能源自多种因素,例如Pd金属中心对富电子芳基的更高耐受性以及温度对这些相互作用的复杂影响。与镍体系的观察结果相呼应,Pd2在所有测试温度下均优于Pd3,尤其是当取代基为含硫的更富电子基团时,链增长受到的抑制更为明显(见图7a)。值得注意的是,随着温度的升高,所有钯催化剂的活性普遍增强,其中Pd1的活性增幅最为显著。这一提升很可能是由于在较高温度下,乙烯插入的能量壁垒有所降低(如图7a所示)。与镍体系的结果相类似,钯体系中的金属-π相互作用对所生成的聚乙烯的分子量产生了显著影响。钯催化乙烯聚合所得聚乙烯的分子量遵循Pd1>Pd2>Pd3的顺序(如图7b所示)。值得注意的是,由Pd3催化得到的聚乙烯分子量明显低于其他两种催化剂,差距超过一个数量级(请见图7b)。这一发现进一步印证了含硫二苯并环庚基取代基对钯催化中心的深远影响。与镍体系相一致的是,随着温度的升高,所有催化剂产生的聚乙烯分子量均有所下降,这主要归因于链转移与链增长比率的降低。然而,与镍体系不同的是,在钯体系中,仅有含硫的二苯并环庚基取代基对降低聚乙烯的支化度具有显著影响(请见图7c)。这可能是因为柔性的、富电子的含硫二苯并环庚基取代基与钯催化中心之间的相互作用更为有效。对聚乙烯进行13C NMR谱分析揭示了其支化结构主要由长链支化构成(如图8所示),从而验证了这些聚合物在室温下于THF中的溶解度。综上所述,在钯体系中,含氧的二苯并环庚基取代基在促进链转移和抑制链行走方面效果不佳,这可以归因于钯中心对富电子环境具有一定的耐受性。图7. 30 ℃和50 ℃下,Pd1-Pd3产生聚乙烯的产量(a)、分子量(b)和支化度(c)比较图8. 在30 ℃下,Pd2产生的支化聚乙烯的13C NMR谱图分析丙烯酸甲酯,作为一种广泛使用的单体,在乙烯共聚反应中占据着举足轻重的地位。它常被用作评估α-二亚胺钯催化剂共聚性能的基准物质。同时,丙烯酸甲酯在极性官能化聚乙烯的合成过程中也扮演着重要角色。本研究发现,与均聚活性相比,钯催化剂在共聚反应中的活性显著降低。这一现象主要是由于丙烯酸甲酯插入后形成了稳定的环状中间体。与钯催化的乙烯均聚反应类似,通过Pd3催化的乙烯与丙烯酸甲酯的共聚反应所生成的乙烯-丙烯酸甲酯共聚物,其分子量明显低于由Pd1和Pd2催化得到的共聚物,同时其支化度也较低(见图9)。这些观察结果表明,在乙烯-丙烯酸甲酯共聚过程中,含硫的二苯并环庚基取代基同样起到了抑制链行走并促进链转移的作用。然而,一个引人注目的现象是,Pd3的共聚活性与Pd1相当,且显著高于Pd2(参见图9a)。尽管其根本原因仍有待深入探究,但这可能与含硫二苯并环庚基取代基在促进稳定环中间体开环方面的作用有关。值得注意的是,在多数情况下,由Pd1和Pd2催化的共聚物中,丙烯酸甲酯的插入比例超过了Pd3。这一差异的具体原因目前尚不明确,但可能与二苯并环庚基取代基和丙烯酸甲酯单体之间的Pd-π相互作用的特性密切相关。图4.9 在1.0和2.0 mol/L丙烯酸甲酯浓度下,用Pd1-Pd3生成的共聚物的产量(a)、插入比(b)、分子量(c)和支化度(d)的比较综上所述,本研究利用α-二亚胺配体,成功设计并合成了一系列具有单边杂原子二苯并环庚基取代基的镍和钯催化剂。在镍催化的乙烯聚合反应中,我们发现杂原子二苯并环庚基镍催化剂的活性较低,所生成的聚乙烯相较于二苯并环庚基镍催化剂,具有更低的分子量和支化度。特别值得一提的是,含硫取代的二苯并环庚基镍催化剂的活性更低,且其产生的聚乙烯分子量和支化度均低于含氧取代的二苯并环庚基镍催化剂。在钯催化的乙烯聚合中,我们也观察到类似的规律:杂原子的二苯并环庚基钯催化剂相较于对应的二苯并环庚基钯催化剂,更倾向于生成低分子量和低支化度的聚乙烯。尤为引人注目的是,在钯体系中,含硫的二苯并环庚基取代基对聚合反应的影响更为显著。在杂原子二苯并环庚基钯催化剂的共聚反应中,也呈现出类似的趋势,显著特点是能够生成低分子量且低支化度的共聚物。我们将这些观察结果归因于催化中心上相邻基团间的金属-π相互作用。具体来说,在乙烯的(共)聚合过程中,与富电子的杂原子二苯并环庚基取代基相关的增强的金属-π相互作用,有助于促进链转移并抑制链行走。特别是在镍和钯体系中,含硫的二苯并环庚基取代基因其强给电子能力和较柔性的电子结构,显著地促进了链转移并抑制了链行走。本研究不仅为金属催化乙烯聚合体系的设计和优化提供了宝贵的参考,更展示了通过合理选择配体和取代基来精准调节聚乙烯性能的可行性。该研究成果发表在《J. Catal.》(DOI:10.1016/j.jcat.2024.115550),陆卫青和丁北航为该论文共同第一作者。
免责声明:本内容来自腾讯平台创作者,不代表腾讯新闻或腾讯网的观点和立场。
 在线客服
在线客服